Comparative and Longitudinal Connectomics
Micha Sam Brickman Raredon
2020-05-13
04 Comparative and Longitudinal Connectomics.RmdIn this vignette, we show how to compare connectomics across multiple single-cell datasets, indepedent of node parcellation. The functions below are able to compare cell-cell signaling topologies across conditions, timepoints, and even across tissues/organs. For demonstration, we use the muscle regeneration dataset from DeMicheli et al. 2020.
Load and Format data
GSE143437_DeMicheli_MuSCatlas_metadata <- read.delim("GSE143437_DeMicheli_MuSCatlas_metadata.txt") GSE143437_DeMicheli_MuSCatlas_rawdata <- read.delim("GSE143437_DeMicheli_MuSCatlas_rawdata.txt") raw.data <- GSE143437_DeMicheli_MuSCatlas_rawdata meta.data <- GSE143437_DeMicheli_MuSCatlas_metadata rm(GSE143437_DeMicheli_MuSCatlas_rawdata) rm(GSE143437_DeMicheli_MuSCatlas_metadata) # Reformat rownames raw.data.2 <- raw.data[,-1] rownames(raw.data.2) <- raw.data[,1] meta.data.2 <- meta.data[,-1] rownames(meta.data.2) <- meta.data[,1]
Data Structure and Processing
This dataset contains 10x data across 4 timepoints during a muscle injury and regeneration model. Here, we convert the raw counts to a Seurat object, split the data by injury timepoint, and then make the corresponding x4 tissue connectomes.
# Create Seurat Object musc <- CreateSeuratObject(counts = raw.data.2) musc <- AddMetaData(musc,metadata = meta.data.2) Idents(musc) <- musc[['cell_annotation']] # Identify ligands and receptors which have mapped in the dataset: connectome.genes <- union(Connectome::ncomms8866_mouse$Ligand.ApprovedSymbol,Connectome::ncomms8866_mouse$Receptor.ApprovedSymbol) genes <- connectome.genes[connectome.genes %in% rownames(musc)] # Split by timepoint data <- SplitObject(musc, split.by = 'injury') for (i in 1:length(data)){ data[[i]] <- ScaleData(data[[i]]) } # Normalize, Scale, and create Connectome: musc.con <- list() for (i in 1:length(data)){ data[[i]] <- NormalizeData(data[[i]]) data[[i]] <- ScaleData(data[[i]],features = genes) musc.con[[i]] <- CreateConnectome(data[[i]],species = 'mouse',p.values = F) } names(musc.con) <- names(data)
Note that differential connectomics cannot easily be performed here, because the data has been parcellated into different nodes at different time points.
| Day 0 | Day 2 | Day 5 | Day 7 | |
|---|---|---|---|---|
| Anti-inflammatory macrophages | 0 | 0 | 4839 | 792 |
| B/T/NK cells | 50 | 0 | 450 | 147 |
| Endothelial | 2678 | 317 | 549 | 1353 |
| FAPs | 2425 | 583 | 2591 | 3161 |
| Mature skeletal muscle | 566 | 0 | 176 | 159 |
| Monocytes/Macrophages/Platelets | 0 | 2749 | 3174 | 0 |
| MuSCs and progenitors | 156 | 0 | 1641 | 786 |
| Neural/Glial/Schwann cells | 266 | 0 | 0 | 148 |
| Pro-inflammatory macrophages | 0 | 1315 | 0 | 0 |
| Resident Macrophages/APCs | 164 | 560 | 820 | 304 |
| Smooth muscle cells | 509 | 0 | 0 | 387 |
| Tenocytes | 214 | 0 | 0 | 409 |
This is okay! This is exactly the use-case we have designed CompareCentrality for.
Comparing mechanistic centrality across disparate datasets
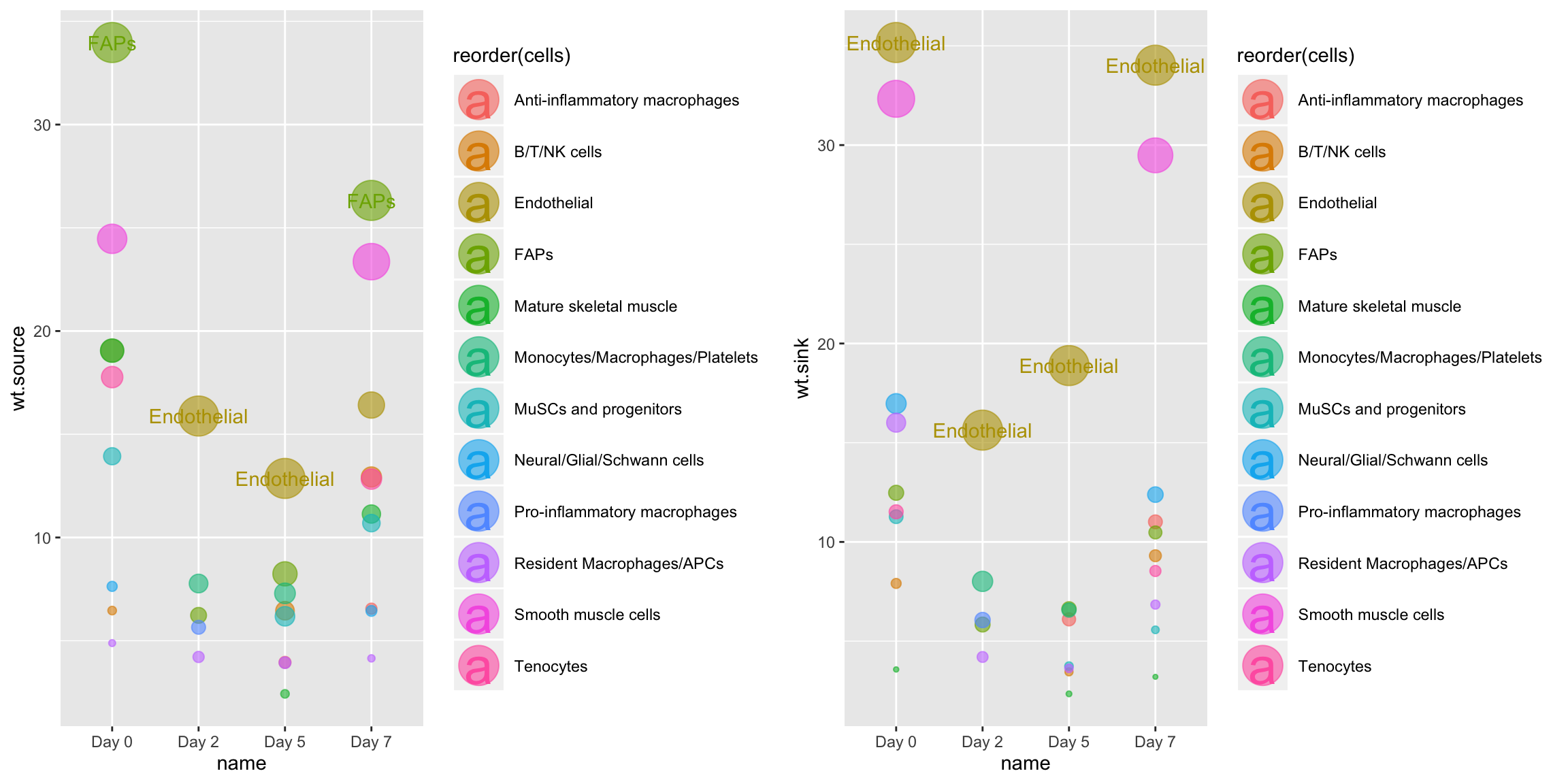
CompareCentrality is similar to Centrality, but instead of grouping in the input network into sub-networks based by a requested group.by term, it analyzes a list of input networks and compares their signaling topologies side-by-side. This allows comprehensive ranking of signal producers and receivers within a chosen network across multiple tissue systems (in this case, x4 timepoints following injury). Here, we analyze the entire VEGF signaling family network across all timepoints:
# Comparative centrality CompareCentrality(musc.con, weight.attribute = 'weight_norm', modes.include = 'VEGF')
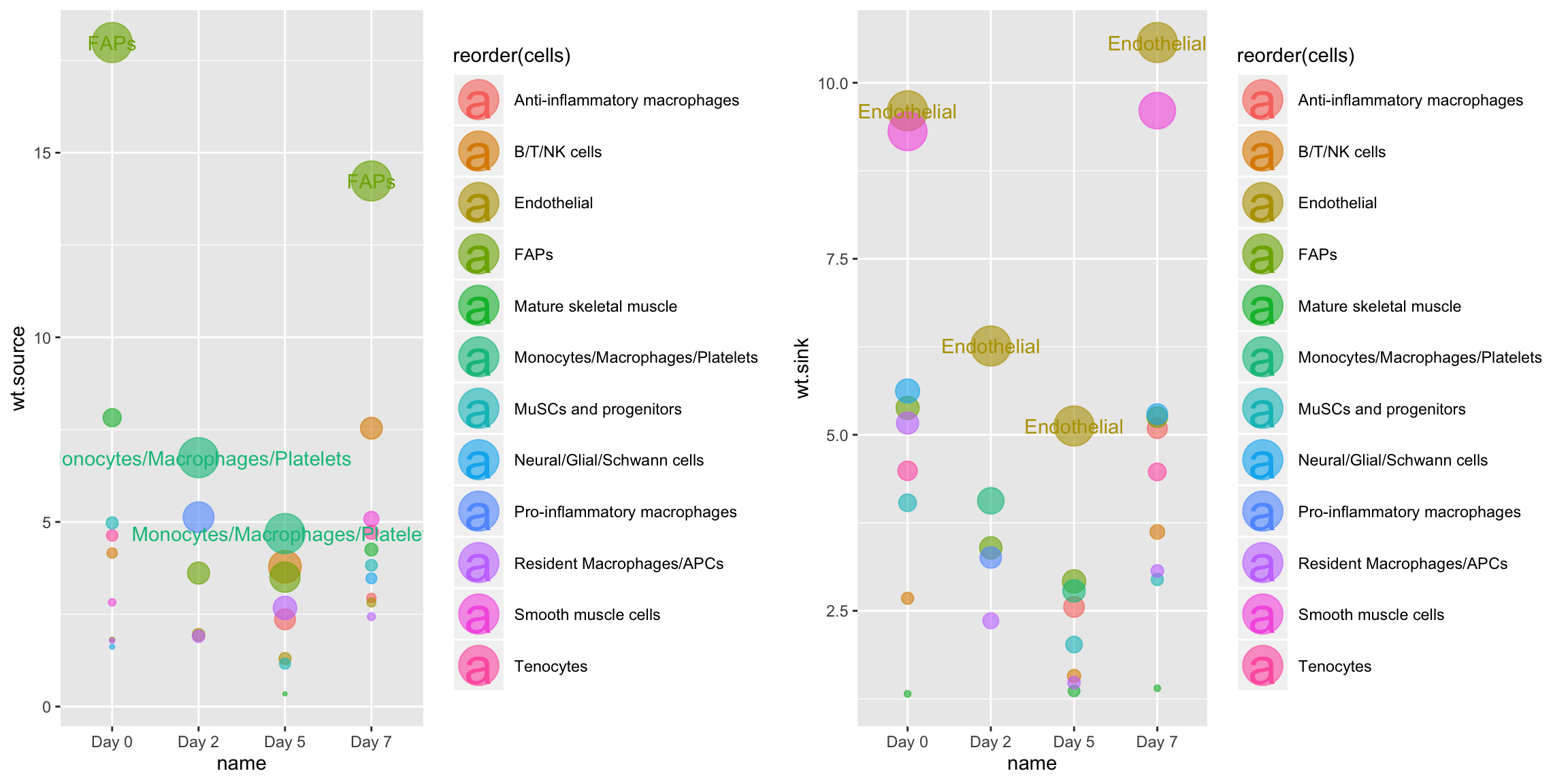
We can also use this function to look at network centrality for only those edges involving a specific gene:
CompareCentrality(musc.con, weight.attribute = 'weight_norm', #min.pct = 0.1, features = 'Vegfa')
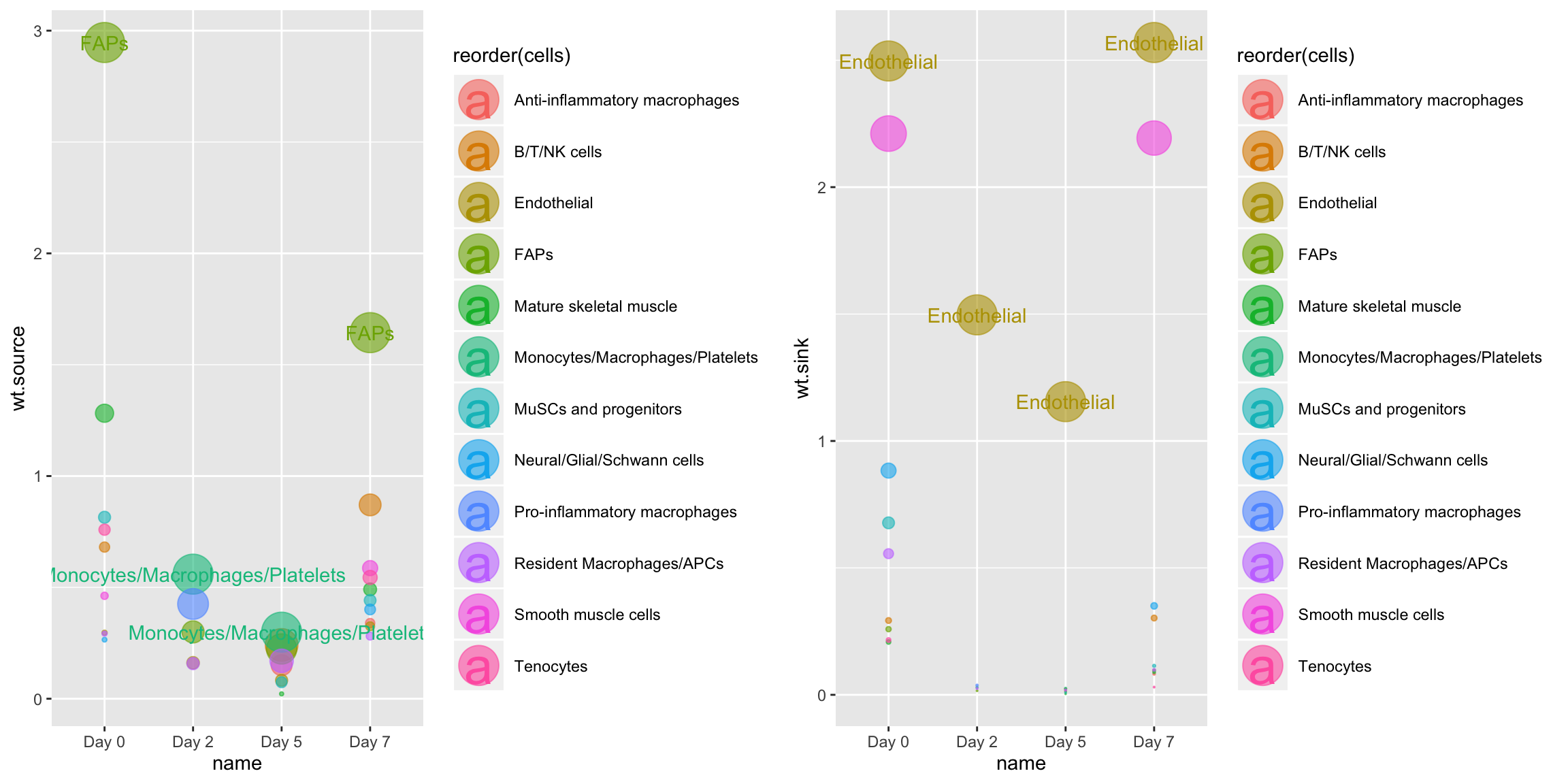
Or, we can use CompareCentrality to quantify signaling centrality across all timepoints for only a specific mechanism:
CompareCentrality(musc.con, weight.attribute = 'weight_norm', #min.pct = 0.1, mechanisms.include = 'Vegfa - Flt1')
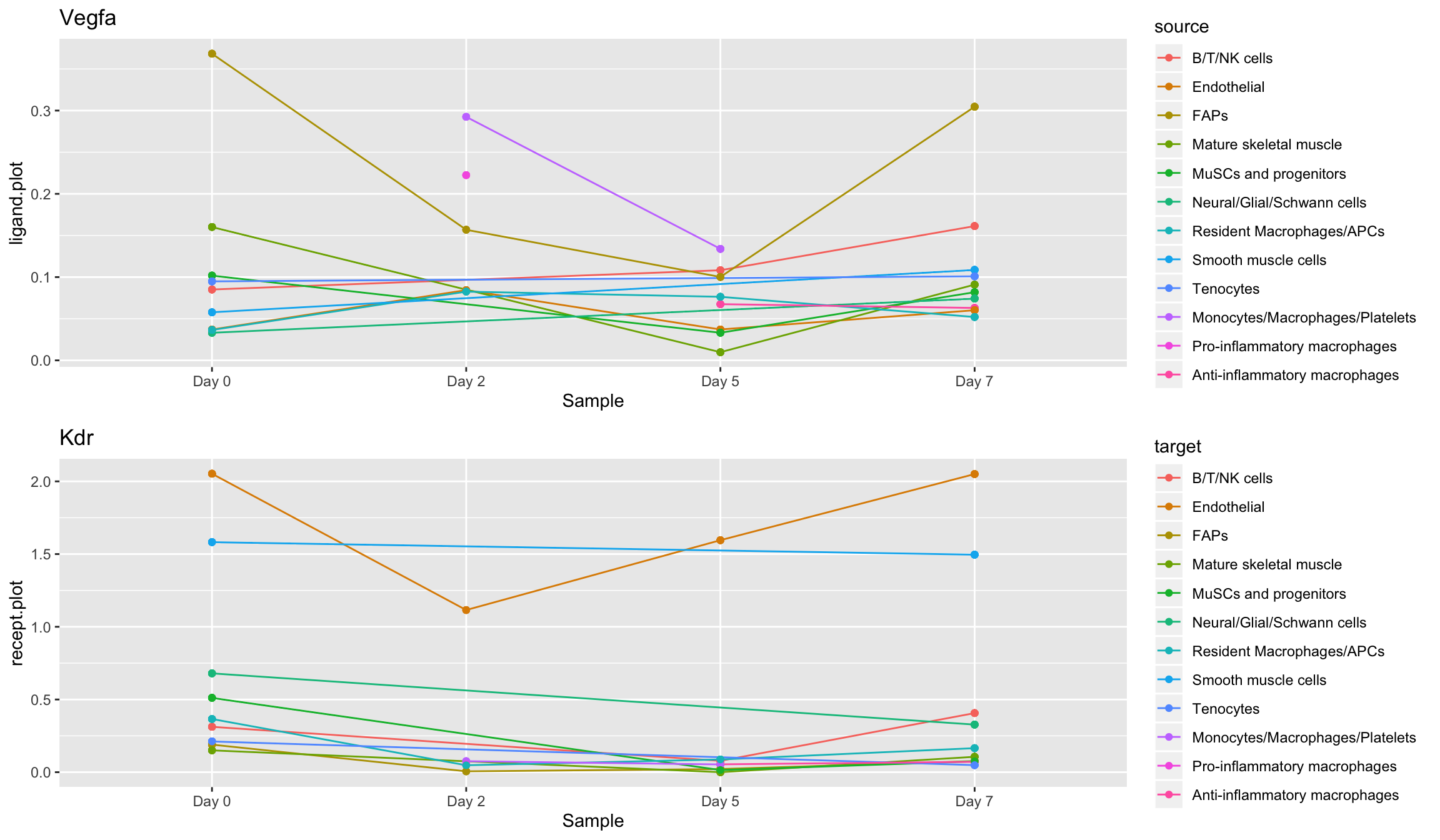
Longitudinal Ligand - Receptor Contours
It should be noted that the Connectome data frames lend themselves well to longitudinal ggplotting which allow observation of trends over time. Since we already have a dataset loaded which captures 4 timepoints in a post-injury regeneration model, let’s take a look at how certain ligand - receptor topologies change over time.
Define a longitudinal plotting function
First, let’s define a simple function to format our data
Longitudinal <- function(con.list,LOI,ROI,use.scaled = F,...){ data <- data.frame() for (i in 1:length(con.list)){ con.list[[i]]$sample.name <- names(con.list)[i] data <- rbind(data,con.list[[i]]) } temp <- subset(data,ligand == LOI & receptor == ROI) if(use.scaled == F){ temp$ligand.plot <- temp$ligand.expression temp$recept.plot <- temp$recept.expression }else{ temp$ligand.plot <- temp$ligand.scale temp$recept.plot <- temp$recept.scale } p1 <- ggplot(temp,aes(x = factor(sample.name, levels = names(con.list)), y = ligand.plot,group = source,color = source)) + geom_line() + geom_point() + xlab('Sample') + ggtitle(LOI) p2 <- ggplot(temp,aes(x = factor(sample.name, levels = names(con.list)), y = recept.plot,group = target,color = target)) + geom_line() + geom_point() + xlab('Sample') + ggtitle(ROI) plot_grid(p1,p2,ncol=1) }
Longitudinal plots
Now we’re prepared to observe ligand trends and receptor trends over time, for each cell type which was defined in the original single-cell data. We can see from the following plots that FAP cells only display high levels of Vegfa in their homeostatic states (Day 0 and Day 7), while immediately post-injury and during hearing (Day 2 and Day 5) a Monocyte/Macropahge population arises which appears to temporarily support Vegfa secretion:
Longitudinal(musc.con,LOI = 'Vegfa',ROI = 'Kdr')